
Researchers search for efficient, active substances to combat diseases, often relying on compounds that interact with proteins that trigger physiological actions. Due to the abundance of chemical compounds, this search resembles finding a needle in a haystack. Drug discovery leverages scientific models, and with the advent of AI, machine learning applications, such as graph neural networks (GNNs), have gained prominence.
Graph Neural Networks in Drug Discovery
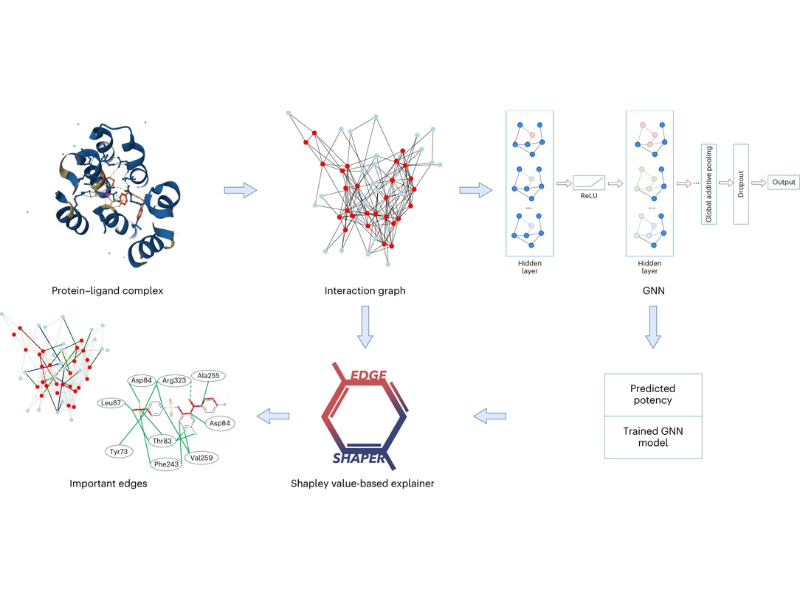
GNNs, like “Graph neural networks,” are crucial in predicting how strongly a molecule binds to a target protein. They are trained using graphs representing complexes formed between proteins and chemical compounds.
However, the inner workings of GNNs remain elusive, prompting researchers to investigate their ability to learn protein-ligand interactions.
Analyzing GNNs and Their Predictions
The study, led by Prof. Dr. Jürgen Bajorath and colleagues, scrutinized six GNN architectures. The researchers employed the “EdgeSHAPer” method to determine whether these GNNs effectively learned the essential interactions between a compound and a protein to predict binding strength accurately.
The Clever Hans Effect in GNN Predictions
The findings revealed that most GNNs primarily focused on ligands and failed to learn crucial protein-drug interactions. Instead, they tended to ‘remember’ chemically similar molecules encountered during training, regardless of the target protein.
This phenomenon, likened to the “Clever Hans effect,” raises questions about the reliability of GNN predictions in drug discovery.
Implications for Drug Discovery Research
The research suggests that GNN predictions may be overrated, as more straightforward methods and chemical knowledge can yield equivalent results.
However, the study also highlights potential avenues for improvement, particularly in GNNs that tend to learn more interactions with increased compound potency.
Looking Ahead: Explainable AI in Drug Discovery
Prof. Bajorath emphasizes the importance of understanding AI models’ predictions. The development of analysis tools, such as EdgeSHAPer, aims to shed light on the black box of AI models, fostering transparency and advancing the field of Explainable AI.
The Lamarr Institute, where Prof. Bajorath is a PI and Chair of AI in the Life Sciences, anticipates exciting developments in Explainable AI, contributing to the broader landscape of artificial intelligence research.
Read the original article on Nature Machine Intelligence.
Source: Mastropietro, A. et al, Learning characteristics of graph neural networks predicting protein–ligand affinities, Nature Machine Intelligence (2023). DOI: 10.1038/s42256-023-00756-9. www.nature.com/articles/s42256-023-00756-9.
Read more: Improving AI Intuition in the Discovery of New Medicines.